By Rizma Khan1, Umaira Zakir2, Faizan-ul-Hassan Naqvi2
AFFILIATIONS:
- Department of Molecular Genetics, Dr. Ziauddin Hospital, Karachi.
- Department of Biochemistry, University of Karachi, Karachi, Pakistan.
ABSTRACT
Essential epigenetic mechanism i.e., DNA methylation in humans is being continuously acknowledged as a hallmark of various cancer. Hypo-methylation and CpG hyper methylation considered early events in cancer development hence; their understanding will provide us new tools for diagnosis. Hypo-methylation of repetitive sequences associated with genomic instability and may cause changes in the local chromatin environment and disrupt gene expression that contributes to carcinoma development. Additionally, hyper methylation of the promoter region of genes, including p15, p16, RASSF1A, accumulates during cancer development, which can influence the process of angiogenesis, DNA repair, regulation of cell cycle, apoptosis as well as tumour cell invasion. The methylation process mediated via DNA methyltransferases; hence, the variation in these enzymes may lead to hepatocellular carcinoma (HCC). Oncogenes activation and tumour suppressor genes inactivation during the growth and development of HCC, promotor hyper-methylation and hypo-methylation are the activaters. This review summarized recent DNA methylation information and role of aberrant methylation in cancer progression by using different research papers from NCBI between 2016-2020, that would be helpful in the diagnosis and treatment of hepatocellular carcinoma.
Keywords: DNA Methylation; CpG Island; Gene Expression; Hypo-Methylation; Hyper- Methylation; Review.
Hepatocellular carcinoma (HCC) is a multistage carcinogenetic process, includes the development of genetic and epigenetic alterations in various genes that later become a major reason for different oncogenes activation and tumour suppressor genes inactivation1, such as hyper-methylation induce silencing of numerous critical genes are an early event in HCC2. Epigenetic modifications are hallmarks of cancer; understanding of factors associated with the Cancer progression will have a significant impact on the management and treatment of the disease3. Previous studies suggested that the accumulation of methyl group on DNA is a highly dynamic process susceptible for pharmacological therapy and is an important therapeutic target4. Post-genomics era is now based on epigenetic modifications that are prone to suppression of genes due to association with methyltransferases5 activity and expression of these epigenetic enzymes including DNA and histone methyltransferases altered in hepatocarcinogenesis6,7.
Previously it has been reported that human epigenetic markers are involved in the maintenance of DNA methyltransferase activity and its overexpression has been identified in several human malignancies8,10. DNA methylation is a frequently researched process essential for the advancement of almost all cancer11, 12. Tumours often have a lower methylation level of genomic DNA and hyper-methylation of CpG islands13-15. Oncogenes activation and tumour suppressor genes inactivation during the growth and advancement of human hepatocellular carcinoma (HCC), promotor hyper-methylation and hypo-methylation are the most dominating triggers16-18.
Aberrant DNA methylation is one of the epigenetic changes that have a potential role in hepatocellular carcinoma (HCC). Genome-wide analysis revealed that in both cases of loss and gain in DNA methylation are frequent measures that happened in HCC. DNA methylation can change the activity of DNA without any change in the sequence6. A normal process occurs in all cells but if it gets abrupt that result in various types of cancer such as cervical cancer and gastric cancer means both global genome wide hypo-methylation and hyper-methylation linked with cancer, autoimmune and neurological diseases15, 18.
Aberrant Methylation and Status of CpG Islands
Methylation of nucleotide bases originated from different taxonomic groups, in which N6-methyladenine (6mA), N4-methylcytosine (4mC), and 5-methylcytosine (5mC) been clearly described. Among these 4mC and 6mA delimited to firm eukaryotes but for all prokaryotes18-21. While the epigenetic modification mainly occurs in eukaryotic DNA is 5mC21. In mammals, 5mC plays a crucial role in X-inactivation, gene silencing as well as genomic imprinting21.
It has been recognizing that for a certain period promoter CpG Island (CGIs) methylation does not occur unintentionally. A random border from the starting point of transcription displays those sixty percent of human genes subordinate by CpG islands22. Nevertheless, un-methylated CpG are not uniformly distributed, however, they typically assembled in the regions called CGIs23. CGIs have a DNA sequence of ~1-kb short stretch CpG di-nucleotides24. Approximately thirty thousand CGIs found in the human genome and observed that CGIs in promoter region usually remain un-methylated in the normal cells25. In the promotor region of mammals, approximately 45000 CGIs exist. The results of promoter CGIs methylation incite an enduring X chromosome inactivation, transcriptional silencing of the related genes, genomic imprinting26, and inactivation of genes. That play a role in tumour suppression like Growth Arrest and DNA Damage Inducible Alpha (GADD45A), Ras association domain family 1 isoform A (RASSF1A), cyclin-dependent kinase inhibitor 2B (CDKN2B or p16), Secreted Frizzled Related Protein 1 (SFRP1), these epigenetic aberrations occur throughout HCC development27.
Other epigenetic alteration such as hypo-methylation of repetitive sequence that is linked with genomic instability is also recognized event in HCC27. In HCC rat model, initiation of the multistage carcinogenesis occurs by epigenetic alterations, such as hyper-methylation in the promoter CGIs of tumour suppressor genes p16, RASSF1A, suppressor of cytokine signalling 1 (SOCS-1), connexin 26 (Cx26), and Cadherin-1 (CDH1). These changes occur by disrupting the balance between cell proliferation and apoptosis that are the basic pro tumorigenic event in hepatocellular carcinoma28. These findings suggest that the production of abnormal methylation signature in healthy tissues or an epigenetic domain is predisposed to cancer development29.
DNA Methylation in Transcriptional Start Sites, Gene Body and its Repetitive Sequence
On the other hand, transcriptional factors and their control in gene expression are extensively under the observation of scientists30. Hence, to start gene transcription, a promoter region would be willingly available to factors associated with transcription and further controlling elements31. In the normal cells of human genome, approximately 60% of the gene transcriptional start sites (TSSs) contain CGIs and are normally un-methylated. CGIs methylation in TSSs is commonly, invoked as a mechanism for transcriptional silencing and can directly prevent binding of the transcription factor to its normal sites. For instance, PEG3-DMR sequence methylation in vivo inhibits the transcription factor YY1 binding, consequence in the maternal allele silencing32. While in paternal allele, YY1 can resourcefully drag to un-methylated sequence of PEG3-DMR and form an appropriate assembly for transcription33. Whereas methylated CpG recruits methyl-CpG-binding domain (MBD) proteins that attract repressor complexes, ultimately modification in histone proteins occurs that leads to further compressed heterochromatin structure which is contrasting toward euchromatin required for transcription34 (Figure 1).
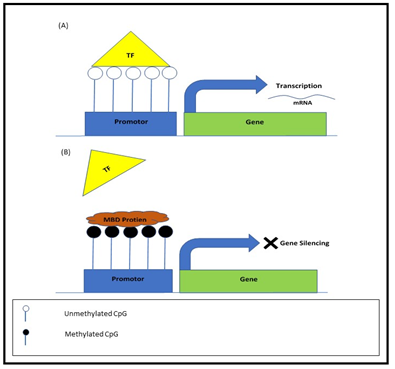
Figure 1: Transcriptional mechanism of DNA methylation in TSSs. (a): The CpG Island is un-methylated and allows binding of transcription factor (TF) to un-methylated binding sites and thus allowing transcription. (b): The CpG Island is a methylated recruiting methyl-CpG-binding domain (MBD) that restricting access of transcription factors to gene promoter and results in gene silencing.
Gene bodies’ methylation is essential mechanism for the management of promoter usage and contains CGIs, but most of them are CpG-poor and methylated heavily. Research reveals that an increase in the methylation of the gene body might associate with gene activation35. The research investigates that methylation among introns and exons were necessary for the regulation of the splicing process35.
Repetitive sequences mainly consist of interspersed repeats as well as tandem repeats, these sequences of the genome in normal somatic cells methylated densely and vital for genomic stability and normal gene expression, de-methylation of the repetitive sequence also referred to as global hypo-methylation, leads to genomic instability and frequently observed in tumour and/ or surrogate tissues36.
Hypo versus Hyper-methylation and Hepatocellular Carcinoma
Hypo-methylation is a modification, which predominantly grasped in solid tumours and is a sign of a progressive malignancy37. Repetitive genomic regions, which comprise of Alu-type repetitive regions and long interspersed nuclear element, various retroviruses classes and centromeric satellite repeats, encompass equal to 55 percent of the human genome, are sensitive to 5-methylcytosine loss38. The intragenic areas and introns in DNA sequence and promoter CGI of oncogene cause global hypo-methylation39. That linked to chromosomal instability, genomic imprinting loss and recurrence of transposable elements in the earliest step of HCC. Moreover, hypo-methylation of satellite alpha repetitive sequences results in increased expression of satellite alpha transcript (SAT), which induces abnormal segregation of chromosomes, leads to chromosomal instability40. In human HCC various hypo-methylated tumour-promoting genes identified, these groups of genes activated in tumours by hypo-methylation.
Furthermore, hyper-methylation can influence the process of DNA repair, angiogenesis, and regulation of the cell cycle, tumour cell invasion and apoptosis. In humans, cancer genes inactivated by hyper-methylation that is essential for the sustainability of stem cell appearances during cell regeneration41. Additionally, hyper-methylation can prime to the suppression of genes that are necessary to the common anti-tumour process. Aberrant methylation of CGIs of various genes responsible for tumour suppression, including RASSF1a, Glutathione S-transferase Pi 1 (GSTP1), Adenomatous polyposis coli (APC), and SOCS-1, cause transcriptional silencing of these genes, that is the reason for the clinical outcome of HCC42.
Commonly Studied Hyper-Methylated Marker Genes in Hepatocellular Carcinoma
The down regulation of tumour suppressor genes via promoter methylation within CGIs is one of the key molecular mechanisms in HCC43. Research on alteration in DNA methylation of tumour suppressor gene, p16 plays a vital role to understand the mechanisms of oncogenesis44. Its promoter hyper-methylation thoroughly related to the damage of p16 appearance that is responsible for HCC development45. Hyper-methylation of common tumour suppressor genes in HCC listed in Table 1.
Table 1: Targeted genes biological process, functions and pathways.
| Gene Symbols | PTM Sites
(Phosphorylated) |
Biological Process | Molecular Function | Pathways |
| RASSF1A | S2-p, R18-m1, K21-ac, R36-m1, T38, T43, S135-p, K142-ub, S179, S182, S183,K185-ub,S188-p, R198-m1, S201,T206-p, S207-p,K216-ub, S222-p, T224-p, K236-ub,K245-ub, Y259-p,K262-ub, L264,K281-ub,K326-ub | · Cell cycle arrest;
· Ras protein signal transduction; · Regulation of microtubule cytoskeleton organization |
· Protein binding;
· Zinc ion binding |
· Ras signaling pathway
· Hippo signaling pathway · Pathways in cancer · Micro RNAs in cancer · Bladder cancer · Non-small cell lung cancer
|
| GADD45A | T2-p, T2-n-ub, K12-ub, K12-sm, K18-ub, K29-ub, S32-p, Y41-p | · Activation of MAPKKK activity;
· apoptotic process; · cell cycle arrest; · cellular response to ionizing radiation; · cellular response to mechanical stimulus; · centrosome cycle; · DNA damage response, · signal transduction by p53 class mediator resulting in cell cycle arrest; · DNA repair; mitotic cell cycle arrest; · negative regulation of protein kinase activity; · positive regulation of apoptotic process; · positive regulation of JNK cascade; · positive regulation of p38MAPK cascade; · positive regulation of reactive oxygen species metabolic process; · regulation of cyclin-dependent protein serine/threonine kinase activity; |
· Kinase binding; protein binding;
· promoter sequence-specific DNA binding; · RNA polymerase II core · protein; · protein N-terminus binding homo/ heterodimerization activity |
· G2/M DNA Damage Checkpoint,
· T-Cell Receptor Signaling · MAPK signaling pathway · FOXO signaling pathway · p53 signaling pathway · Apoptosis · Transcriptional misregulation in cancer · Gastric cancer |
| CDH1 | T66-p, Y68-p, S70-p, T211-p, T217-ga, T330-p, T576-p, T599-p, N637, Y663-p, K738-ub, T748-p, Y753-p, Y754-p, Y755-p, S770-p, T790-p, S793-p, Y797-p, S838-p, S840-p, S844-p, S846, S847-p, S850-p, S851,S853-p, K871-ub, Y876-p | · Adherens junction organization;
· cell-cell adhesion; · cellular response to indole-3-methanol; cellular response to lithium ion; · entry of bacterium into host cell; · extracellular matrix organization; · homophilic cell adhesion via plasma membrane adhesion molecules; · negative regulation of cell migration; · negative regulation of cell-cell adhesion; · neuron projection development; pituitary gland development; · positive regulation of protein import into nucleus; · positive regulation of transcription, DNA-templated; · protein localization to plasma membrane; · regulation of gene expression substance |
· Ankyrin binding;
· beta-catenin binding; · cell adhesion molecule binding; gamma- catenin binding; · GTPase activating protein binding; · identical protein binding; protein binding |
· Rap1 signaling pathway
· Apelin signaling pathway · Hippo signaling pathway · Cell adhesion molecules (CAMs) · Pathways in cancer · Bladder cancer · Gastric cancer |
Gene families of Growth arrest and DNA damage-inducible 45 (Gadd45) comprises of 03 members, Gadd45α, 5β, and 45γ. They remain concerned in numerous trials such as cell proliferation, apoptosis, epigenetic modification and DNA repair46,47. Up until now, various studies show that the key mechanism concerned with the down expression may be because of methylation at the promoter region in HCC48. They act as trauma devices also called stress sensors, arbitrating the multifaceted interchange of physical connections with proteins, responsible for cell cycle and stress stimuli regulation including Cdc2/cyclin B, PCNA, JNK stress response kinases P21, P3849. In addition, active de-methylation associated with MBD4 and TDG has directly under the surveillance of GADD4550. Furthermore, GSTP1, tumour suppressor gene defend cells from loss of DNA reconciled by various electrophiles are hyper-methylated in HCC51.
CDH1 is another well-known tumour suppressor gene that has an important role in the care of the environment and cell polarity. Inactivation of this gene allied with tumour progression, cell invasion and metastasis52. Aberrant methylation in CGIs is accountable for the loss of CDH1 expression in cancer also LOH (loss of heterozygosity) of this gene linked with hyper-methylation of CpGs53. Thus, correction of DNA methylation eminence in HCC cancer cell may prime to recrudescence of CDH1. Previously it revealed that, snail gene regulate via DNA methylation and directly inhibits the expression of E-cadherin, which provokes EMT-MET transition in HCC54. Expression of CDH1 not only repressed by snail during EMT transition but also aggravates the expression of mesenchymal markers and deviate the cell phenotype55. This gene is also responsible for cell death by removal of persistence aspects like ERK, PI3K and suppression of checkpoints of cell cycle like p21, which is cell cycle inhibitor in either G2/M or G1/S phase56.
Another gene RASSF1A is present in the 3p21.3 region has crucial key role in cell cycle control, apoptosis, motility and cellular adhesion57. Hyper-methylation of RASS1A is the root cause of epigenetic silencing of RASSF1A in many tumours including HCC, kidney cancer, lung cancer, endometrial carcinoma, and breast cancer that result in the loss of gene expression58-60.
DNA Methylation Maintenance and Inheritance
Methylation of DNA has vital role in storage of biological information that catalysed by key enzymes DNA methyltransferase. These are necessary for mammalian development and shift the -CH3 group from S-adenosyl-L-methionine that is methyl universal donor to the cytosine in fifth position61,62, possess methyltransferase activity and responsible for normal human development63. They encode the de novo methyltransferase to create methylation signature in DNA sequences, on the other hand mainly acted as maintenance methyltransferase and throughout replication regulate DNA methylation by replicating the methylation signature of the parental strand to the daughter DNA strand64,65. Instead of its methylation activity, these enzymes have a potent role in DNA damage response66. Research related to DNA damage response demonstrates that their activity as genome guardian in contradiction of double strand breaks of DNA in colon cancer and B cells (cell lines) 66.
The research revealed that knockout of methyltransferase induces premature lethality at 9.5 embryonic days, subsequently after several developmental retardations namely rostral neural tube defects and growth deficit such as growth impairement67. It has a role in embryonic development and knockout induces early embryonic lethality68, 69. Unlike many other, they may not exhibit any intrinsic methylation catalytically function, but according to Bourchis et al. methyltransferases was the key to the development of maternal genomic imprinting70.
Furthermore, play an essential part in genomic integrity; interruption in these may lead to chromosomal instability and tumorigenesis. Research demonstrates that they are essential for silencing many sequence types, particularly inactive X chromosome genes, imprinted genes and transposable elements71 and inhibition of these genes are required to maintain chromosome stability. Research evident a strong association between DNA methylation and DNA repair mechanism in various types of cancer 66.
DNA Methyltransferases Expression in HCC
Methyltransferases involve in mRNA-increased expression from normal liver to HCC and numerous other cancer72. In knockout mice of glycine N-methyltransferase gene, hypo-methylation and abnormal expression of these enzymes reported in the development of different HCC stages73. In the HCC cell line of human, the suppression of DNA methyltransferases reduces proliferation and re-established Phosphatase and tensin homolog (PTEN) gene, that is a central tumour suppressor in HCC, suggested that PTEN seems to be the novel target74. According to the study of Fan et al. Metastasis suppressor protein 1 (MTSS1) is a target of this particular enzyme that has a role in tumour inhibition in HCC was suppressed by overexpression through a methylation-independent mechanism75.
Tian et al. found that Hepatitis B X-interacting (HBX) protein has a role in the epigenetic modification of hepaocarcinogeneis76. They observed that HBx decrease the expression of insulin-like growth factor-3 by de novo methylation76. Moreover, it causes hypo-methylation of satellite 2 repeat sequences by decreasing the expression of methyltransferases76. Studies have shown that Micro RNA has also a role in the control of altered methylation patterns in HCC caused by HBV. In hepatocellular carcinoma (cell lines), increased expression of microRNA 152 (miR-152) leads to the reduction in expression, contributed to a decline in methylation levels and increased the methylation status of tumour suppressor genes77. It also reported that HBx downregulated the miR-101 by targeting transferase enzyme that leads to abnormal DNA methylation78.
Methylation disruption is the molecular mechanisms responsible for the pathogenesis of hepatocarcinogenesis; hyper-methylation of CpG islands of the promoter region of genes is a vital mechanism for transcriptionally silencing of tumour suppressor genes in HCC. In addition, hypo methylation of oncogenes is responsible for chromosome instability and reactivation of transposable elements in the earliest step of HCC. Aberrant DNA methylation of CGIs may take part in hepatocarcinogenesis, so it would be beneficial to recognize the aberrant methylation role in HCC that would help to reduce the mortality and morbidity of the disorder by decreasing the abrupt methylation. This review covered the basic concept of methylation and its role in HCC development.
Authors are grateful to Dr. Nadir Naveed Siddiqui, Assistant Professor at the Karachi Institute of Biotechnology and Genetic Engineering (KIBGE) and the University of Karachi for providing the scientific literature.
The authors declare no conflict of interest.
RK conceived the present review. UZ and FN selected the articles and acknowledged valuable material. RK and UZ did the research writing and submission of the review. All authors read and approved the final version of the manuscript.
- Kanda M, Sugimoto H, Kodera Y. Genetic and epigenetic aspects of initiation and progression of hepatocellular carcinoma. World J Gastroenterol. 2015;21(37):10584-10597.
- Anestopoulos I, Voulgaridou GP, Georgakilas AG, Franco R, Pappa A, Panayiotidis MI. Epigenetic therapy as a novel approach in hepatocellular carcinoma. Pharmacol Ther. 2015;145:103-119.
- Cheng J, Wei D, Ji Y, Chen L, Yang L, Li G, et al. Integrative analysis of DNA methylation and gene expression reveals hepatocellular carcinoma-specific diagnostic biomarkers. Genome Med. 2018;10(1):1-1.
- Huang S, He X. The role of microRNAs in liver cancer progression. Br J Cancer. 2011;104(2):235-240.
- Kaneda A, Matsusaka K, Aburatani H, Fukayama M. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012;72(14):3445-3450.
- Lambert MP, Ancey PB, Esposti DD, Cros MP, Sklias A, Scoazec JY, et al. Aberrant DNA methylation of imprinted loci in hepatocellular carcinoma and after in vitro exposure to common risk factors. Clin Epigenetics. 2015;7(1):1-3.
- Marra M, Sordelli IM, Lombardi A, Lamberti M, Tarantino L, Giudice A, et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: an overview. J Transl Med. 2011;9(1):1-4.
- Balogh J, Victor III D, Asham EH, Burroughs SG, Boktour M, Saharia A, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma. 2016;3: 41-53.
- Zakharia K, Luther CA, Alsabbak H, Roberts LR. Hepatocellular carcinoma: Epidemiology, pathogenesis and surveillance – implications for sub-Saharan Africa. S Afr Med J. 2018; 108(8b):35-40.
- Petrick JL, Braunlin M, Laversanne M, Valery PC, Bray F, McGlynn KA. International trends in liver cancer incidence, overall and by histologic subtype 1978–2007. Int J Cancer. 2016;139(7):1534-1545.
- Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301-1314.
- Imbeaud S, Ladeiro Y, Zucman-Rossi J. Identification of novel oncogenes and tumor suppressors in hepatocellular carcinoma. Semin Liver Dis. 2010;30(1):75-86.
- Paska AV, Hudler P. Aberrant methylation patterns in cancer: a clinical view.Biochem Med (Zagreb). 2015;25(2):161-176.
- Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 2015;161(4):879-892.
- Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, et al. N6-methyladenine DNA modification in Drosophila. Cell. 2015;161(4):893-906.
- Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6(5):1-22.
- Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel). 2017;8(6):1-20.
- Feng P, Chen W, Lin H. Prediction of CpG island methylation status by integrating DNA physicochemical properties. Genom. 2014;104(4):229-233.
- Long HK, King HW, Patient RK, Odom DT, Klose RJ. Protection of CpG islands from DNA methylation is DNA-encoded and evolutionarily conserved. Nucleic Acids Res. 2016;44(14):6693-6706.
- Jeziorska DM, Murray RJS, De Gobbi M, Gaentzsch R, Garrick D, Ayyub H, et al. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc Natl Acad Sci USA. 2017;114(36): E7526-E7535.
- Ferreira HJ, Esteller CpG islands in cancer: Heads, tails, and sides. Methods Mol Biol. 2018;1766:49-80.
- Gao S, Wang K. DNA methylation in liver diseases. World J Clin Infect Dis. 2016;4(4):41-49.
- Pogribny IP, Muskhelishvili L, Tryndyak VP, Beland FA. The role of epigenetic events in genotoxic hepatocarcinogenesis induced by 2- Mutat Res. 2011;722(2):106-113.
- Hattori N, Ushijima T. Epigenetic impact of infection on carcinogenesis: mechanisms and applications. Genome Med. 2016;8(1):1-3.
- Sullivan AM, Arsovski AA, Lempe J, Bubb KL, Weirauch MT, Sabo PJ, et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014;8(6):2015-2030.
- Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, et al. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015;12(7):1184-1195.
- He H, Ye A, Perera BPU, Kim J. YY1’s role in the peg3 imprinted domain. Sci Rep. 2017;7(1):1-1.
- Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nat. 2010;464(7291):1082-1086.
- Li L, Chen BF, Chan WY. An epigenetic regulator: Methyl-CpG-binding domain protein 1 (MBD1). Int J Mol Sci. 2015; 16(3):5125-5140.
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Sci. 2007;315(5815):1141-1143.
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nat. 2011;479(7371):74-79.
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nat. 2010;466(7303):253-257.
- Maumus F, QuesnevilleDeep investigation of Arabidopsis thaliana junk DNA reveals a continuum between repetitive elements and genomic dark matter. PLoS One. 2014; 9(4): 1-14.
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15(1):1-7.
- Zheng Y, Joyce BT, Liu L, Zhang Z, Kibbe WA, Zhang W, et al. Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res. 2017; 45(15):8697-8611.
- Zheng Y, Hlady RA, Joyce BT, Robertson KD, He C, Nannini DR, et al. DNA methylation of individual repetitive elements in hepatitis C virus infection-induced hepatocellular carcinoma. Clin Epigenetics. 2019;11(1):1-3.
- Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Reply to Haffner et al.: DNA hypomethylation renders tumors more immunogenic. Proc Natl Acad Sci. 2018;115(37):E8583-E8584.
- Lisanti S, Omar WA, Tomaszewski B, De-Prins S, Jacobs G, Koppen G, et al. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PloS One. 2013; 8(11): 1-11.
- Lee ST, Wiemels JL. Genome-wide CpG island methylation and intergenic demethylation propensities vary among different tumour sites. Nucleic Acids Res. 2016; 44(3):1105-1117.
- Nishida N, Kudo M, Nishimura T, Arizumi T, Takita M, Kitai S, et al. Unique association between global DNA hypomethylation and chromosomal alterations in human hepatocellular carcinoma. PloS One. 2013;8(9):1-10.
- IchidaK, Suzuki K, Fukui T, Takayama Y, Kakizawa N, Watanabe F, et al. Overexpression of satellite alpha transcripts leads to chromosomal instability via segregation errors at specific chromosomes. Int J Oncol. 2018;52(5):1685-1693.
- Anestopoulos I, Voulgaridou GP, Georgakilas AG, Franco R, Pappa A, Panayiotidis MI. Epigenetic therapy as a novel approach in hepatocellular carcinoma. Pharmacol Ther. 2015; 145:103-119.
- Pfeifer GP. Defining driver DNA methylation changes in human cancer. Int J Mol Sci. 2018;19(4):1-13.
- Araujo OC, Rosa AS, Fernandes A, Niel C, Villela-Nogueira CA, Pannain V, et al. RASSF1A and DOK1 promoter methylation levels in hepatocellular carcinoma, cirrhotic and non-cirrhotic liver, and correlation with liver cancer in Brazilian patients. PLoS One. 2016;11(4):1-10.
- Li Y, Cai Y, Chen H, Mao L. Clinical significance and association of GSTP1 hypermethylation with hepatocellular carcinoma: A meta-analysis. J Can Res Ther. 2018; 14: S486-S489.
- Zhang W, He H, Zang M, Wu Q, Zhao H, Lu LL, et al. Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterol. 2017;153(1):249-262.
- Zhang X, Wang J, Cheng J, Ding S, Li M, Sun S, et al. An integrated analysis of SOCS1 down-regulation in HBV infection-related hepatocellular carcinoma. J Viral Hepat. 2014; 21(4):264-271.
- Jia W, Yu T, Cao X, An Q, Yang H. Clinical effect of DAPK promoter methylation in gastric cancer: A systematic meta-analysis. Med. 2016;95(43):1-8.
- Ullah F, Khan T, Ali N, Malik FA, Kayani MA, Shah ST, et al. Promoter methylation status modulate the expression of tumour suppressor (RbL2/p130) gene in breast cancer. PLoS One. 2015;10(8): 1-17.
- Toiyama Y, Okugawa Y, Goel A. DNA methylation and microRNA biomarkers for noninvasive detection of gastric and colorectal cancer. Biochem Biophys Res Commun. 2014; 455(1-2):43-57.
- Lv X, Ye G, Zhang X, Huang T. p16 Methylation was associated with the development, age, hepatic viruses infection of hepatocellular carcinoma, and p16 expression had a poor survival: A systematic meta-analysis (PRISMA). Med. 2017;96(38):1-12.
- Liu LQ, Tian FJ, Xiong Y, Zhao Y, Song JB. Gadd45a gene silencing by RNAi promotes cell proliferation and inhibits apoptosis and senescence in skin squamous cell carcinoma through the p53 signalling pathway. J Cell Physiol. 2018; 233(9):7424-7434.
- Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hofman B. Gadd45 stress sensors in malignancy and leukemia. Crit Rev Oncog. 2011; 16(1-2):129-140.
- Liang Z, Liu R, Zhang H, Zhang S, Hu X, Tan J, et al. GADD45 proteins inhibit HIV-1 replication through specifc suppression of HIV-1 transcription. Virol. 2016; 493:1-11.
- Niehrs C, Schafer A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012; 22(4):220-227.
- Qiao CY, Li F, Teng Y, Zhao J, Hu N, Fan YC, et al. Aberrant GSTP1 promoter methylation predicts poor prognosis of acute-on-chronic hepatitis B pre-liver failure. Clin Exp Med. 2018; 18(1):51-62.
- Zhu C, Feng X, Ye G, Huang T. Meta-analysis of possible role of cadherin gene methylation in evolution and prognosis of hepatocellular carcinoma with a PRISMA guideline. Med (Baltimore).2017; 96(16):1-8.
- Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci.2007; 120(18):3163-3172.
- Kawai Y, Sakano S, Suehiro Y, Okada T, Korenaga Y, Hara T, et al. Methylation level of the RASSF1A promoter is an independent prognostic factor for clear-cell renal cell carcinoma. Ann Oncol.2010; 21(8):1612-1617.
- Wang J, Wang B, Chen X, Bi J. The prognostic value of RASSF1A promoter hypermethylation in non-small cell lung carcinoma: a systematic review and meta-analysis. 2011; 32(3):411-116.
- Cui D, Xu X. DNA methyltransferases, DNA methylation, and age-associated cognitive function. Int J Mol Sci. 2018;19(5):1-16.
- Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018; 19(2):81-92.
- Ren W, Gao L, Song J. Structural basis of DNMT1 and DNMT3A-mediated DNA methylation. Genes. 2018; 9(12):1-20.
- Jurkowska RZ, Jeltsch A. Silencing of gene expression by targeted DNA methylation: Concepts and approaches. Methods Mol Biol. 2010; 649:149-161.
- Sharma S, De Carvalho DD, Jeong S, Jones PA, Liang G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet. 2011;7(2): 1-14.
- Hayashi K, Hishikawa A, Itoh H. DNA damage repair and DNA methylation in the kidney. Am J Nephrol. 2019;50(2):81-91.
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3(9):662-673.
- Nowialis P, Lopusna K, Opavska J, Haney SL, Abraham A, Sheng P, et al. Catalytically inactive Dnmt3b rescues mouse embryonic development by accessory and repressive functions. Nat Commun. 2019; 10(1):1-6.
- Li Y, Zhang Z, Chen J, Liu W, Lai W, Liu B, et al. Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nat. 2018;564(7734):136-140.
- Bourchis D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Sci. 2001;294(5551): 2536-2539.
- Niller HH, Nagy-Demcsák A, Minarovits J. DNA methylation in eukaryotes: Regulation and function. Cell Ecophysiol Microbe Hydrocarb Lipid Interact. 2018:509-550.
- Honeywell RJ, Sarkisjan D, Kristensen MH, de Klerk DJ, Peters GJ. DNA methyltransferases expression in normal tissues and various human cancer cell lines, xenografts and tumors. Nucleosides Nucleotides Nucleic Acids. 2018;37(12):696-708.
- Szparecki G, Ilczuk T, Wolosz D, Otto W, Gornicka B. The expression of DNA methyltransferase DNMT3a in classical and fibrolamellar hepatocellular carcinoma. J Clin Exp Pathol. 2016;6(3):1-4.
- Zhao Z, Wu Q, Cheng J, Qiu X, Zhang J, Fan H. Depletion of DNMT3A suppressed cell proliferation and restored PTEN in hepatocellular carcinoma cell. J Biomed Biotechnol. 2010;2010:1-11.
- Fan H, Chen L, Zhang F, Quan Y, Su X, Qiu X, et al. MTSS1, a novel target of DNA methyltransferase 3B, functions as a tumor suppressor in hepatocellular carcinoma. Oncogene. 2012;31(18):2298-2308.
- Tian Y, Yang W, Song J, Wu Y, Ni B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol Cell Biol. 2013;33(15):2810-2816.
- El-Araby RE, Khalifa MA, Zoheiry MM, Zahran MY, Rady MI, Ibrahim RA, et al. The interaction between microRNA-152 and DNA methyltransferase-1 as an epigenetic prognostic biomarker in HCV-induced liver cirrhosis and HCC patients. Cancer Gene Ther. 2020;27(6):486-497.
- Wei X, Xiang T, Ren G, Tan C, Liu R, Xu X, et al. miR-101 is down-regulated by the hepatitis B virus x protein and induces aberrant DNA methylation by targeting DNA methyltransferase 3A. Cell Signal. 2013;25(2): 439-446.