By Sundus Nasim1, Dua Azim1, Farheen Malik1, Faryal Tahir1, Zehra Ashraf 2, Abdul Qayyum3
AFFLIATIONS:
- Dow Medical College, Dow University of Health Sciences, Karachi, Pakistan.
- Jinnah University for Women, Karachi, Pakistan.
- Department for Oncology, Dr. Ziauddin Hospital, Karachi, Pakistan.
ABSTRACT
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, aggressive type of non-Hodgkin’s lymphoma (NHL) that is mostly diagnosed in elderly patients. Its non-specific clinical presentation (lymphadenopathy, fever, night sweats, weight loss, generalized rash, and hepatosplenomegaly) often results in a delay in the diagnosis. The diagnosis is mainly established based on a detailed clinical evaluation and biopsy findings, and currently, available treatment options include corticosteroids, immunotherapy, and single- or multi-agent chemotherapy. Here, we report a case of a 61-year-old male who presented with complaints of easy fatigability, dyspnea, and fever along with inguinal lymphadenopathy and was diagnosed as a case of AITL. He was given multiple cycles of R-CHOP chemotherapy (Cytoxan, Hydroxyrubicin, Oncovin, Prednisone chemotherapy regimen), which led to tumor eradication. The patient, however, expired due to unknown reasons. The case highlights the major diagnostic modalities and treatment strategies for AITL and sheds light on the poor prognosis of the disease despite adequate management.
Keywords: Angioimmunoblastic Lymphadenopathy; Lymphoma; T-Cell; Non-Hodgkin; Chemotherapy.
Angioimmunoblastic T-cell lymphoma (AITL) is a rare non-Hodgkin’s lymphoma (NHL) in which the follicular T helper (TFH) cells undergo a malignant and aggressive transformation. This particular hematologic malignancy contributes to only 1-2% of all NHLs, and the prevalence of AITL in Pakistan is as low as 0.41%1,2. AITL typically presents in the sixth or seventh decade of life with constitutional symptoms, such as weight loss, fever, night sweats, hepatosplenomegaly, lymphadenopathy, and skin rash3. With a median 5-year survival of 32%, the disease prognosis is poor. These grim statistics could be due to frequent relapses or delay in the diagnosis of AITL, as most patients usually present at an advanced stage1. The present case report aimed to contribute to the knowledge and understanding of AITL’s disease course and the development of post-treatment complications. No evidence of drug toxicity contributing to death was witnessed in this case. Thus, despite treatment, the fatal outcome of this case warrants further research about the exact cause of death.
A 61-year-old male with type 2 diabetes mellitus presented to the emergency department of Dr. Ziauddin Hospital, Karachi, with complaints of easy fatigability, dyspnea, and low-grade fever accompanied by night sweats. The patient also reported significant weight loss. He had no significant history of cardiovascular disease, smoking, illicit drug intake, or chemical exposure; family history was non-contributory.
Physical examination revealed bilateral wheezes over the chest, unilateral left inguinal lymphadenopathy, and splenomegaly. Initially, pulmonary tuberculosis (TB) with dissemination to inguinal lymph node was suspected; however, the hard consistency of the lymph nodes pointed towards an underlying malignancy.
Complete blood count (CBC) with peripheral blood smear showed microcytic hypochromic anemia, bicytopenia, and hyper-segmented neutrophils. The Mantoux test was negative, ruling out Tuberculosis. Laboratory investigations during the initial hospital visit showed an elevation in the serum alkaline phosphatase (ALP) [160 IU/L, N=39-117 IU/L], beta-2 microglobulin (6.5 mg/L, N=<2.4), and lactate dehydrogenase (276 U/L, N=135-225 U/L). A chest x-ray revealed bilateral pulmonary infiltrates (Figure 1). Bone marrow aspiration was negative for extranodal involvement.
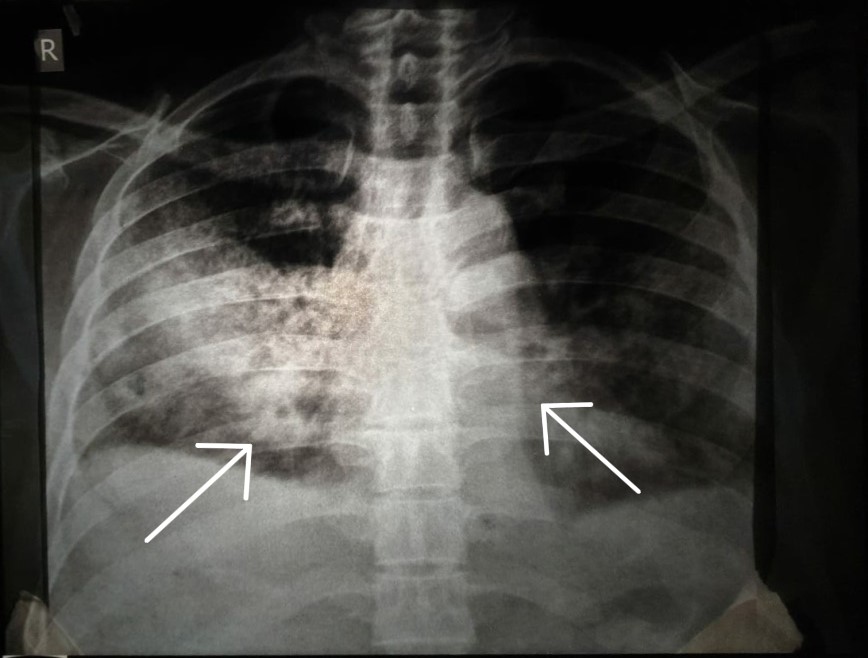
Figure 1: Chest X-ray showing bilateral pulmonary infiltrates.
Inguinal lymph node biopsy exhibited few scattered large cells with abundant cytoplasm, pleomorphic vesicular nuclei and prominent nucleoli. Immunohistochemical stains were positive for CD3 and CD4 in small cells, which highlighted a population of T lymphocytes with elevated Ki-67 (Mib-1). Additionally, stains for CD20 were positive in a few large cells; scattered B-cells also showed positive staining for CD10, CD21, PAX-5, BCL-6, and PD-1. It was thus decided to diagnose this as a case of AITL.
Pre-chemotherapy full-body positron emission tomography/computed tomography scan (PET/CT) showed hypermetabolic lymphadenopathy involving the cervical, supraclavicular, axillary, mediastinal, hilar, abdominal, retroperitoneal, pelvic, and inguinal regions bilaterally (Figure 2A). Axial PET/CT scan of mediastinum revealed hypermetabolic areas in the lungs, along with evidence of bilateral pleural effusion and mediastinal lymphadenopathy (Figure 3A). Overall, the imaging findings were suggestive of a diffuse lymphoproliferative disorder. Subsequently, the patient was started on R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy, and he attained complete remission after two cycles of R-CHOP therapy. Post-chemotherapy full-body PET/CT scan showed no metabolic activity in the previously noted areas (Figure 2B). The axial PET/CT imaging of mediastinum (Figure 3B) showed no evidence of effusion as well as reduced metabolic activity. These imaging were indicative of a satisfactory response to the prescribed therapeutic regimen; hence, the patient was discharged.
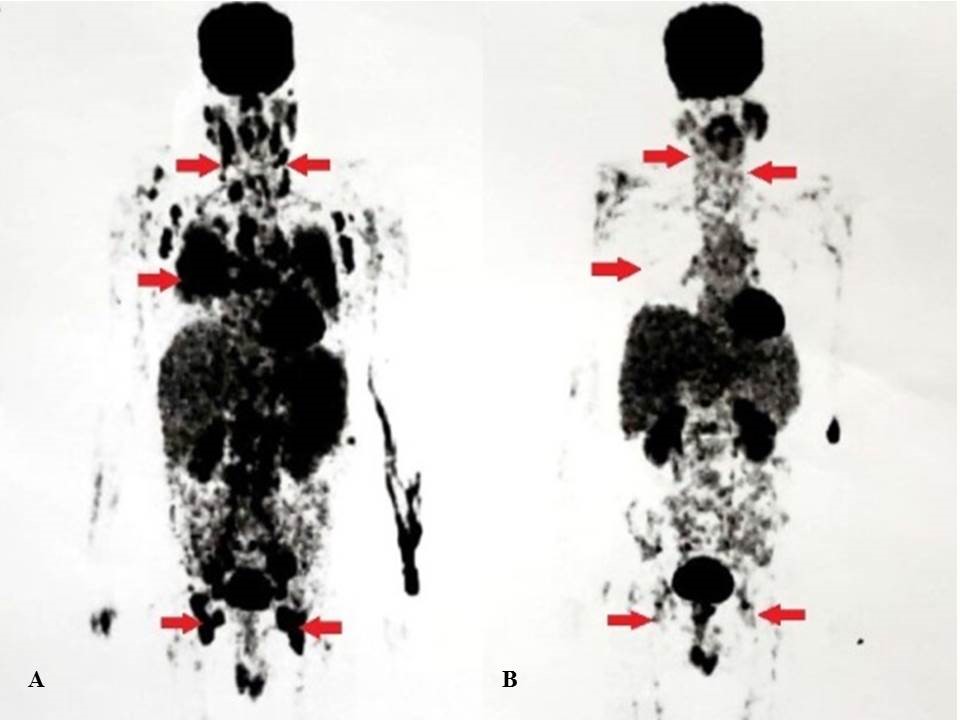
Figure 2: (A) Pre-chemotherapy positron emission tomography/computed tomography scan showing diffuse hypermetabolic activity; (B) Post-chemotherapy positron emission tomography/computed tomography scan showing reduced metabolic activity in previously noted areas.
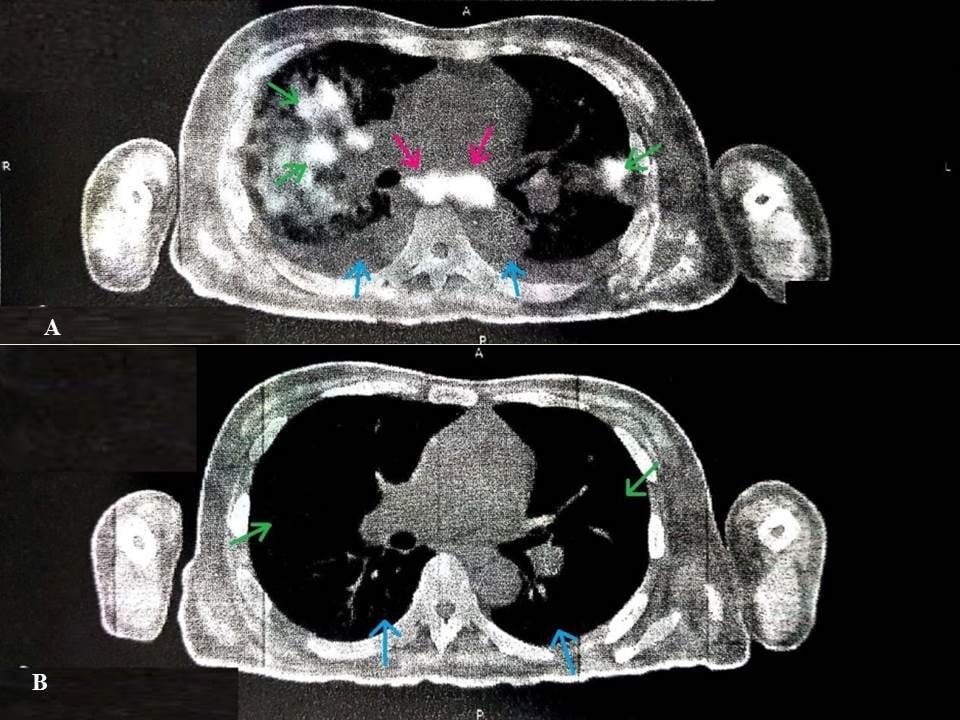
Figure 3: (A) Pre-chemotherapy positron emission tomography/computed tomography scan showing multiple hypermetabolic lesions in lungs (green arrows), bilateral pleural effusion (blue arrows) and mediastinal lymphadenopathy (pink arrows); (B) Post-chemotherapy positron emission tomography/computed tomography scan showing reduced metabolic activity in previously noted areas (green arrows) with no evidence of pleural effusion (blue arrows).
After two weeks of remission, the patient again reported to the ED with complaints of fever and dyspnea. Laboratory investigation showed pancytopenia with significant leukopenia (1.1×109/L, N=4.0-10) and thrombocytopenia (40×109/L, N=150-440). After obtaining blood cultures, the patient received intravenous fluids and empiric antibiotics. Platelets were transfused for his worsening thrombocytopenia, and extensive blood workup was performed to exclude platelet transfusion reaction. Although blood cultures came out negative, the patient continued to deteriorate and eventually developed multiple organ failure (MOF). The patient also developed respiratory distress requiring endotracheal intubation and mechanical ventilation. Despite all possible efforts, the patient expired a week after readmission. The reason for sudden death remains unidentified, despite thorough imaging and laboratory investigations.
AITL is a rare hematological malignancy accounting for approximately 1-2% of NHLs, and almost 20% of all peripheral T-cell lymphoma (PTCL) cases diagnosed per year. It classically presents in the elderly with a mean age of sixty-five years. Unlike the other PTCL subtypes, the incidence of AITL is more common in Europe (28.7%) relative to Asia (17.9%) 1. Diagnosis of AITL depends on the clinical picture, laboratory investigations, and biopsy report. Imaging techniques such as computerized tomography (CT) scan, magnetic resonance imaging (MRI), and positron emission tomography (PET) scan may help in the diagnosis of AITL3.
The initial clinical presentation is usually ambiguous, which includes generalized lymphadenopathy, skin rash, hepatosplenomegaly, and development of B symptoms such as fever, night sweats, and weight loss. Moreover, autoimmune dysfunction is often the cause of many AITL symptoms, such as arthritis, vasculitis, thyroid abnormalities, autoimmune hemolytic anemia, and thrombocytopenia; these can easily misdirect the diagnosis towards an autoimmune disorder. Thus, prompt diagnosis of AITL is a significant challenge. Literature suggests that cutaneous manifestations, notably skin rash, are present in many cases. This rash resembles the morbilliform rash of measles, but erythematous and maculopapular appearances are not uncommon3. However, our patient was devoid of any cutaneous involvement. Furthermore, AITL may also spread to extra nodal sites that typically include bone marrow, spleen, lungs, and skin4. Nearly 70% of patients will have bone marrow involvement in the course of the disease1. While the patient did present with splenomegaly and pulmonary infiltrates, there was no bone marrow involvement in this case.
Apart from anemia and thrombocytopenia, laboratory investigations also show evidence of hypergammaglobulinemia, a positive Coombs test, elevated serum LDH level, and erythrocyte sedimentation rate (ESR) 1. On an immunophenotypic analysis, neoplastic T-cells expressed CD3, CD4, and the TFH cell markers, which included CD10, CXCL13, and PD-1. The expression of CD10, BCL6, CXCL13, and PD1 represents an essential adjunct in the diagnosis of AITL, which helps in distinguishing AITL from other PTCLs. The consistent expression of CXCL13 has enhanced diagnostic confidence. Although the expression of these markers is variable, CXCL13 tends to be more specific relative to CD10 1. The pathologic markers expressed in this case were CD3, CD4, CD10, CXCL13, PD-1, and BCL-6. In addition, studies suggest that AITL is often associated with Epstein Barr virus (EBV) positive B-lymphocytes and can mimic viral illness3. This could be the result of a reactivation of EBV due to extreme immunosuppression. However, in this patient, the B cells were EBV negative.
The treatment of AITL includes corticosteroids, chemotherapy, radiotherapy, and stem cell transplantation1,3. Traditionally, both single-agent and combination chemotherapeutic regimens such as CHOP have been used. To this day, CHOP therapy remains the treatment of choice in patients with AITL1. Despite various trials, the addition of drugs like rituximab, alemtuzumab, bevacizumab, and belinostat to the CHOP regimen did not result in any therapeutic benefit5. In this case, CHOP therapy combined with rituximab (R-CHOP) was the first-line treatment. Following two cycles of R-CHOP therapy, the patient went into complete remission, indicating the efficacy of the said treatment modality.
The majority of the cases of AITL are complicated by superimposed infections rather than progressive lymphoma, since the affected individuals may develop immunosuppression due to both the disease and the chemotherapy treatment3. Thus, they are more prone to infections that can potentially cause severe, life-threatening complications and, consequently, death. Disease-specific death is more associated with older age and advanced stages of the disease (stage III and IV) 6.
Additionally, several studies have also identified toxicities secondary to the treatment as a common cause of mortality in patients with AITL. The main adverse effects of CHOP chemotherapy include immunosuppression, infections, cardiotoxicity, and hematological toxicities such as neutropenia and thrombocytopenia5. In this case, we did note hematological toxicities following chemotherapy; rituximab (anti-CD20 monoclonal antibody), in particular, has reported side effects of severe infusion-related reactions and thrombocytopenia requiring hospitalization. Apart from hemorrhagic cystitis (ruled out via urinalysis); the side effects of cyclophosphamide, such as immune suppression and alopecia, were also evident in this case7. Doxorubicin can also cause significant cardiotoxicity that might prove fatal8, but electrocardiography (ECG) and echocardiogram of this patient did not show any evidence of cardiotoxic side effects8. The exact cause of death in AITL is unknown; however, evidence reveals respiratory failure, cardiac failure, and infections due to immunosuppression may be significant contributors9.
Hence, more research is required to establish the long-term safety and therapeutic efficacy of the aforementioned treatment strategies for patients with AITL. The potential success in treating AITL effectively and preventing its fatal complications depends on a thorough understanding of the disease pathophysiology and in developing improved therapeutic strategies with more tolerable side effects by conducting large-scale clinical trials.
A careful study of this case led us to conclude that the fatal outcome could be attributed either to the malignancy itself or to the side effects of the chemotherapy. Research that is more clinical is required to reduce the diagnostic delay of AITL and to devise better therapeutic strategies.
The authors would like to acknowledge the Oncology Department of the Dr. Ziauddin Hospital for facilitating the study.
The authors declare no conflict of interest.
Patient’s family have been informed regarding the study and written consent was taken.
All authors contributed equally in this case study.
- Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood. 2017;129(9):1095-1102.
- Shahid R, Gulzar R, Avesi L, Hassan S, Danish F, Mirza T. Immunohistochemical profile of hodgkin and non-hodgkin lymphoma. J Coll Physicians Surg Pak. 2016;26(2):103-107.
- Iannitto E, Ferreri AJ, Minardi V, Tripodo C, Kreipe HH. Angioimmunoblastic T-cell lymphoma. Crit Rev Oncol Hematol. 2008;68(3):264-271.
- Mukherjee T, Dutta R, Pramanik S. Aggressive angioimmunoblastic T cell lymphomas (AITL) with soft tissue extranodal mass varied histopathological patterns with peripheral blood, bone marrow, and splenic involvement and review of literature. Indian J Surg Oncol. 2018;9(1):11-14.
- Broccoli A, Zinzani PL. Angioimmunoblastic T-cell lymphoma. Hematol Oncol Clin North Am. 2017;31(2):223-238.
- Ermann DA, Vardell VA, Silberstein PT. Angioimmunoblastic T-cell lymphoma: patient characteristics and survival outcomes. 2019; 134 (Supplement_1):2194.
- Webb H, Jaureguiberry G, Dufek S, Tullus K, Bockenhauer D. Cyclophosphamide and rituximab in frequently relapsing/steroid-dependent nephrotic syndrome. Pediatr Nephrol. 2016;31(4):589-594.
- McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. 2017;31(1):63-75.
- Dong H, Jin N, Zhang H, Fan L, Zhan X, Li X. Rapid and fatal progression of Epstein-Bar virus-associated paracortical hyperplasia with T-cell oligoclones to overt angioimmunoblastic T-cell lymphoma: a case report. Int J Clin Exp Med. 2017;10(8):12889-12897.
This is an open-access article distributed under the terms of the CreativeCommons Attribution License (CC BY) 4.0 https://creativecommons.org/licenses/by/4.0/