By Fatima Jehangir1, Rabia Faisal2, Arwa Giani3, Zainab Aziz4
AFFILIATIONS:
- Department of Family Medicine, Dr. Ziauddin University Hospital, Karachi, Pakistan.
ABSTRACT
Congenital myopathies (CMs) are a diverse group of rare inherited muscle disorders characterized by muscle weakness and hypotonia. Very few studies have outlined the cases of CM in Pakistani immigrant families. The following is a case of a premature male neonate who was born with global hypotonia, cryptorchidism and scoliosis. He was vitally stable at birth, but his respiratory function deteriorated over time and was on ventilatory support. Chest X-rays suggested atelectasis while lab findings revealed deranged arterial blood gas (ABG) and abnormal complete blood count (CBC) levels. Creatine kinase levels were normal however; electrocardiogram (ECG) showed atrial defect and right ventricle and pulmonary artery hypertension. Nerve conduction studies (NCS) and electromyography (EMG) results were suggestive of Myopathy. The patient succumbed to respiratory failure 12 days after birth. Due to the low incidence of CMs and their variable presentation, it is easy to misdiagnose and hence, this delays the timely and effective intervention.
Keywords: Congenital Myopathy; Arterial Blood Gases; Electromyography; Nerve Conduction Studies.
Congenital Myopathies (CMs) are a diverse group of rare inherited muscle disorders characterized predominantly by muscle weakness and hypotonia1,2. They usually manifest at birth or later in infancy, and may progressively worsen over time3. They are rare with an estimated incidence of 1:25000 and a prevalence of 1:22,480 in Sweden and 1:135,000 in Northern England. As currently, only supportive treatment is available for these genetic disorders, diagnosing this condition is the key to timely management of the patient3. Herein, we have presented a case of a suspected congenital Myopathy.
A male neonate, born to consanguineous parents, was prematurely delivered at 35 weeks gestation via an emergency C-section. The delivery was carried out due to indications of polyhydramnios and fetal growth restriction. At birth, his weight was recorded as 1.52 kgs (Normal: 2.5-4.5 kgs). He had an Apgar score of four at one minute and nine at five minutes (N: 7-10). His vital signs were within the normal range: heart rate 140/min, respiratory rate 45/min, O2 saturation 99%, temperature 37°C.
He was incubated at five minutes selectively and shifted to NICU where he was kept on patient triggered ventilation /target tidal volume (PTV/TTV) mode for ventilatory support through volume guarantee and assist control. Physical examination findings showed hypotonia, undescended testes and thoracic scoliosis. The neonatal reflexes were intact however. The next day, a chest X-ray was performed due to signs of respiratory distress, which revealed opacification of left hemithorax, suggestive of atelectasis.
Various labs were performed over the course of days for which details are given as well. The arterial blood gases (ABGs) revealed that partial pressure of oxygen (PO2) was low throughout; carbon dioxide (PCO2) remained high and showed CO2 retention. Low pH with poor respiratory efforts and PO2 saturation remained low throughout. Creatine kinase remained normal, which are generally normal in congenital myopathies. Slight fluctuation was observed in random blood sugars but overall, within the normal limits (N: 80-160 mg). Complete blood picture revealed monocytosis. Lymphocytosis, anisocytosis and poikilocytosis were seen in peripheral film. Hyperkalemia was observed over the days. Serum direct bilirubin levels were elevated for which phototherapy was performed while serum calcium levels were low. The transthoracic electrocardiography results depicted aneurysmal interatrial septum, mildly dilated right atrium/right ventricle, moderate to severe right ventricle and pulmonary hypertension. The biventricular systolic function was normal. Electromyography (EMG) and Nerve Conduction Study (NCS) test was performed that suggested findings homogenous with a severe Myopathy involving all the limbs. Active denervation was seen in the right rectus femoris, which hinted at an underlying necrotizing process.
Genetic testing was done, the results of which were received after the neonate’s death. The sample reports revealed a pathogenic variant of uncertain significance in five genes in the selected panel of 113 genes. It also indicated an autosomal recessive nature of the disease. However, the genetic testing report mentioned that these variants’ information was not conclusive. A muscle biopsy could not be performed due to the low weight of the newborn. The baby succumbed to respiratory failure 12 days after birth.
Congenital myopathies (CMs) can be classified into five main forms based on histopathological findings on muscle biopsy and/or genetic characterization. However, this classification is not always easy due to clinical and genetic heterogeneity. Severe cases can present with several complications including respiratory insufficiency. Unfortunately, no cure is available yet and the condition can only be managed through supportive treatments. Anticipatory measures including genetic counseling and prenatal diagnosis can also help improve disease outcomes.
Congenital myopathies are a group of rare inherited muscular disorders defined by muscle weakness and global hypotonia1,2. The incidence is reported to be approximately 1:25000 and about 14% of all cases of neonatal hypotonia are associated with congenital myopathies4. It has an estimated prevalence of 1:22,480 in Sweden and 1:135,000 in Northern England5. To the best of our knowledge, no study has been conducted in Pakistan to estimate the incidence of CM. International studies outlining the cases of CM in patients of Pakistani descent are also scarce6-8.
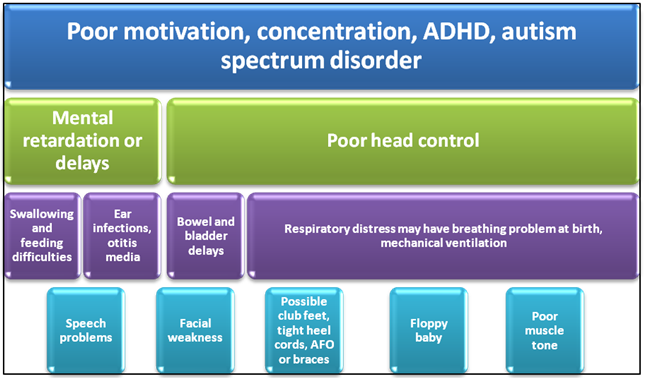
Figure 1: Clinical conditions leading to the congenital myotonic dystrophy (CDM) in neonates.
Congenital myopathies most commonly present in the neonatal period. They can be categorized into five main forms: Nemaline Myopathy (most common type), core Myopathy, Centronuclear Myopathy, congenital fiber-type disproportion Myopathy and myosin storage myopathy3,9. Generally, the disease manifestation is more evident in neonates and in the early-onset cases, which may include reduced fetal movements along with muscle hypotonia at birth, and in the early months of life (Figure 1). In the most severe cases, respiratory insufficiency, severe hypotonia, bulbar dysfunction, and orthopedic abnormalities are present at birth, and the prognosis is unfortunately poor10. The mortality during the first year of life exceeds 10%.6,11.
Several tests are required to confirm the diagnosis of congenital myopathy12. Low muscle mass of the baby and prior EMG testing with needles were the reasons implied for not performing muscle biopsy in our case. Accurate genetic diagnosis of the disease may play a pivotal role in assessing the prognosis, risk and management of the disease13. Until present, the involvement of 20 genes has been specified in the development of CM14; however, the genetic basis in around one third of the patients remains undetermined15. There is no defined correlation between the genotype and the phenotypic expression of the disease. Mutations in different genes may give rise to identical presentation whereas mutations in a single gene may yield distinct or various histopathological findings/ phenotypes2,3,6. Therefore, it can prove challenging for physicians to reach a definitive genetic diagnosis16.
Muscle imaging techniques (magnetic resonance imaging) and muscle ultrasound are used nowadays to identify the genetic basis of the suspected Myopathy thus making it easier to distinguish between the different types17. Genetic testing can be done in situations when a muscle biopsy cannot be carried out (risky biopsy) or excluded any other alternate diagnosis13.
Besides the typical presentation of muscle weakness and hypotonia, many other complications like functional disabilities, cardio-respiratory involvement, orthopedic deformities and motor delay have also been reported in multiple studies6,9. In a review focused on cardiac disorders in Nemaline Myopathy, overall, 35 patients were identified in 29 studies being analyzed18. This study indicated that cardiac involvement is not an infrequent finding but it does not give an inclusive picture of all CMs. Instead, some studies tend to differ and show that heart muscle involvement is still quite variable and rare9. For example, in another retrospective study comprising of 125 patients, only 1 patient had developed cardiomyopathy at the age of 14. In our case too, findings of congenital cardiac abnormalities were determined6.
Amongst the complications, the one which patients of CM are most likely to encounter is respiratory. Overtime, hypoventilation and atelectasis may develop as a result of the gradual weakening of the respiratory muscles19,21. Scoliosis and orthopedic deformities are also generally associated with different forms of myopathies9. Currently, there is no known cure for the disease; only supportive treatments such as assisted breathing, nutritional support, orthopedic corrections and physical, occupational and speech therapies are available9,22. Information regarding the family history and genetic background is vital in evaluating the risk of the disease. Particularly, genetic counseling can prove to be an effective tool in prevention and management of the disease. This motivates families to seek healthy reproductive choices for enhanced family support23. This can especially be put to purpose in high-risk cases of familial types and for couples in consanguineous marriages due to a largely autosomal recessive nature of the disease24.
Due to the low incidence of Congenital Myopathies (CM) and their variable presentation, it is easy to misdiagnose it and hence, delay timely intervention. Clinicians can play a crucial role in laying out an effective management strategy by suspecting and differentiating CM from other similar neuromuscular disorders. This is achievable through application of thorough knowledge and employment of the emerging diagnostic techniques, like ones focused on advanced genetic testing, to help establish the prenatal diagnosis in high-risk pregnancies. This will in turn help to prevent any serious adverse outcomes for the patient and their family.
We would like to acknowledge specifically the patient’s parents for sharing their baby’s data with us. We are also thankful to the Aga Khan University Hospital- Neonatal Intensive Care Unit (NICU).
The authors declare no conflict of interest.
The Ethics Committee of Aga Khan University Hospital, Karachi, Pakistan, approved the study.
The parents of the patient signed informed written consent before using any case information for publishing.
FJ conceived the idea, overall supervised the study and finalized it. RF helped in designing the case report and writing the manuscript. AG and ZA also helped in writing the manuscript.
- Sewry CA, Jimenez-Mallebrera C, Muntoni F. Congenital myopathies. Curr Opin Neurol. 2008;21(5):569-575.
- North KN, Wang CH, Clarke N, Jungbluth H, Vainzof M, Dowling JJ, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord. 2014;24(2):97-116.
- Cassandrini D, Trovato R, Rubegni A, Lenzi S, Fiorillo C, Baldacci J, et al. Congenital myopathies: clinical phenotypes and new diagnostic tools. Ital J Pediatr. 2017;43(1):1-16.
- Tubridy N, Fontaine B, Eymard B. Congenital myopathies and congenital muscular dystrophies. Curr Opin Neurol. 2001;14(5):575-582.
- Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain J Neurol. 2009;132(11):3175-3186.
- Colombo I, Scoto M, Manzur AY, Robb SA, Maggi L, Gowda V, et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurol. 2015;84(1):28-35.
- Vasli N, Harris E, Karamchandani J, Bareke E, Majewski J, Romero NB, et al. Recessive mutations in the kinase ZAK cause a congenital myopathy with fibre type disproportion. Brain J Neurol. 2017;140(1):37-48.
- Koutsopoulos OS, Kretz C, Weller CM, Roux A, Mojzisova H, Bohm J, et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet. 2013;21(6):637-642.
- Wang CH, Dowling JJ, North K, Schroth MK, Sejersen T, Shapiro F, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol. 2012;27(3):363-382.
- Massalska D, Zimowski JG, Bijok J, Kucińska‐Chahwan A, Łusakowska A, Jakiel G, et al. Prenatal diagnosis of congenital myopathies and muscular dystrophies. Clin Genet. 2016;90(3):199-210.
- Romero NB, Monnier N, Viollet L, Cortey A, Chevallay M, Leroy JP, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain J Neurol. 2003;126(11):2341-4349.
- Mah JK, Joseph JT. An overview of congenital myopathies. Continuum (Minneap Minn). 2016;22(6):1932-1953.
- North KN. Clinical approach to the diagnosis of congenital myopathies. Semin Pediatr Neurol. 2011;18(4):216-220.
- Kaplan JC, Hamroun D. The 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord. 2013;23(12):1081-1111.
- Maggi L, Scoto M, Cirak S, Robb SA, Klein A, Lillis S, et al. Congenital myopathies–clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord. 2013;23(3):195-205.
- Cardamone M, Darras BT, Ryan MM. Inherited myopathies and muscular dystrophies. Semin Neurol. 2008;28(2):250-259.
- Fischer D, Herasse M, Ferreiro A, Barragán-Campos HM, Chiras J, Viollet L, et al. Muscle imaging in dominant core myopathies linked or unlinked to the ryanodine receptor 1 gene. Neurol. 2006;67(12):2217-2220.
- Finsterer J, Stollberger C. Review of cardiac disease in nemaline myopathy. Pediatr Neurol. 2015;53(6):473-477.
- Ryan MM, Schnell C, Strickland CD, Shield LK, Morgan G, Iannaccone ST, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. 2001;50(3):312-320.
- Jungbluth H, Sewry C, Brown SC, Manzur AY, Mercuri E, Bushby K, et al. Minicore myopathy in children: a clinical and histopathological study of 19 cases. Neuromuscul Disord. 2000;10(4-5):264-273.
- Khan Y, Heckmatt JZ, Dubowitz V. Sleep studies and supportive ventilatory treatment in patients with congenital muscle disorders. Arch Dis Child. 1996;74(3):195-200.
- Jungbluth H, Ochala J, Treves S, Gautel M. Current and future therapeutic approaches to the congenital myopathies. Semin Cell Dev Biol. 2017;64:191-200.
- Laforgia N, Capozza M, De Cosmo L, Di Mauro A, Baldassarre ME, Mercadante F, et al. A rare case of severe congenital RYR1-associated myopathy. Case Rep Genet. 2018;2018:1-7.
- Corry PC. Consanguinity and prevalence patterns of inherited disease in the UK Pakistani community. Hum Hered. 2014;77(1-4):207-216.