By Afrina Raza1, Shamil Ashraf2, Soofia Nigar3, Itimad Abdelsalam Mohamed Ayed1,4
- Department of Pharmacology, Unaizah College of Medicine, Qassim University, Buraydah-52571, Saudi Arabia.
- Indus Hospital and Health Network, Karachi, Pakistan.
- Department of Anatomy, Dow University of Health Sciences, Karachi, Pakistan.
- Department of Pharmacology, Unaizah College of Medicine, Qassim University and Dermatology and Venerology, Unaizah College of Medicine and Medical Sciences, Qassim University, Buraydah-52571, Saudi Arabia.
DOI: https://doi.org/10.36283/PJMD12-1/003
How to cite: Raza A, Ashraf S, Nigar S, Ayed IAM. Genetic Polymorphism of Thiopurine S-Methyltransferase in Children with Acute Lymphoblastic Leukemia. Pak J Med Dent. 2023;12(1): 5-11. doi: 10.36283/PJMD12-1/003
Background: 6-Mercaptopurine (6-MP), a widely used anti-metabolite for the maintenance phase of childhood acute lymphoblastic leukemia (ALL), has been observed to cause myelotoxicity due to genetic polymorphism of thiopurine methyl transferase (TPMT), one of the drug-metabolizing enzymes. This study aimed to determine the frequency of thiopurine S-methyl transferase (TPMT) polymorphic variants in a group of Pakistani children with acute lymphoblastic leukemia (ALL) using 6-mercaptopurine (6-MP).
Methods: Diagnosed cases (n=100) of ALL, either Pre-B or T Cell, between the ages of 2 to 18 years were randomly selected from OPD of Children Cancer Hospital and National Institute of Child Health (NICH), Karachi. The subjects were under BFM (Berlin Frankfurt Munster) protocol. Expired and relapsed patients (2/100) were excluded. Blood samples were drawn and DNA was extracted from serum to determine the genotype of TPMT (*2, *3a, *3B and *3C) using allele-specific PCR (AS-PCR) and RFLP. PCR assays were done to detect the G238C transversion in TPMT*2 and the G460A and A719G transition in TMPT*3 Alleles.
Results: Polymorphism in TMPT was found 100% (98/98) in ALL patients belonging to a heterozygous group. Out of them, 74.5% of ALL patients showed myelotoxicity. Furthermore, *1/*3B was the most prevalent variant TPMT allele observed with the highest frequency reported at 97%. The variant allele *1/*3A was found in only 3 patients, whereas, TMPT*2 and *3C Alleles were not found.
Conclusion: The majority of our patients displayed a distinct TPMT genetic polymorphism TPMT*1/*3B which is comparatively rare in other parts of the world.
Keywords: 6-Mercaptopurine; Precursor Cell Lymphoblastic Leukemia-Lymphoma; Genetic Polymorphism; Alleles; Pakistan.
6-Mercaptopurine (6-MP) is a purine anti-metabolite that is widely used for the maintenance phase of childhood acute lymphoblastic leukemia (ALL)1; the most common type of cancer in children2. A large number of patients who are using 6-mercaptopurine demonstrate myelotoxicity3, which is attributed to genetic polymorphism of one of the drug-metabolizing enzymes, thiopurine methyl transferase (TPMT)4. It is a cytosolic enzyme involved in the methylation (inactivation) of 6-mercaptopurine and 6-thioguanine5,6. Activation of prodrug 6-MP normally occurs by the action of hypoxanthine-guanine phosphoribosyl transferase (HGPRT) to form thioguanine nucleotide (TGN) and then its final incorporation into DNA to exert its cytotoxic effect7. An alternate pathway of metabolism of this drug includes the inactivation of the drug either to S-methylated metabolites by TPMT7 and 6-thiouric acid by xanthine oxidase (XO)8. Inactivation of the drug ultimately decreases its availability for activation of TGNs all over the body including bone marrow cells. This way it prevents the chances of developing the lethal effect of myelotoxicity.
The activity of TPMT is inherited as an autosomal codominant trait exhibiting TPMT polymorphism9. Worldwide studies show the trimodal distribution in TPMT activity with approximately 89-90%, 10-11% and 0.3% possessing high, intermediate, and low intra-cellular TPMT activity9. Approximately 24 allelic variants of TPMT have been reported10; with a preponderance of TPMT*2, *3A, *3B and *3C being the most common6. All of these variant alleles result from point mutations in the coding sequence or mutations at intron-exon splice sites. The molecular defect in TPMT*2 is G238 C transversion (mutation) that leads to an amino substitution at codon 80 (Ala80 Pro). The TPMT*3A allele contains two different nucleotide transitions (mutations) namely G460 A and A719 G which led to the amino acid substitutions Ala154 Thr and Tyr240 Cys respectively. The other two common variant alleles TPMT*3B and TPMT*3C contain only single nucleotide transitions G460A leading to amino acid substitution Ala154 Thr is present in TPMT*3B allele while transitioning A719 G leading to Amino acid substitution of Tyr – Cys shows TPMT*3C as the polymorphic allele11.
Currently, sufficient data on TPMT genotyping of Pakistani ALL children does not exist. The present study aimed to determine the pattern of TPMT polymorphism in a group of Pakistani ALL patients undergoing treatment with 6 – MP. This TMPT genotyping will discriminate various polymorphic alleles in the group of ALL patients with low and high activity of TPMT. Genotyping of TMPT will help in identifying patients prone to myelosuppression. Hence, children at higher risk of developing myelosuppression will be managed by an adjusted dosage of 6MP. This study was designed to determine the frequency of thiopurine S-methyl transferase (TPMT) polymorphic variants in a group of children of acute lymphoblastic leukemia (ALL) using 6-mercaptopurine (6-MP), anti-metabolite for the maintenance phase of childhood acute lymphoblastic leukemia (ALL), in Pakistan.
One hundred children of ALL ages 2 to 18 years were randomly registered for this case series study from OPD of Children Cancer Hospital and National Institute of Child Health (NICH). Informed consent was taken from their parents/guardians. Before this approval from the hospital ethics review committee was obtained. All the patients were receiving chemotherapy according to BFM protocol. Two patients, one had a relapse and one passed away during the study, were excluded. Five ml of blood was drawn from 98 patients and serum was separated. All sera samples were stored at -800C. DNA was extracted from buffy-coat by using Resin based DNA extraction buffer (BioSweep, Korea). 4.5 µl of the extraction DNA was used for each PCR amplification reaction by adding 12.5 µl of Go-Taq green master mix (Promega, USA).
Allele-specific PCR (AS-PCR) was done for the detection of variants TPMT*2 (G238C), TPMT*3A (G460A and A719G), TPMT*3B (G460A) and TPMT*3C (A719G). the four variant TPMT alleles*2, *3A and *3C were amplified by detection of transversion G238C and transitions G460A and A719G in each patient. Separate PCRs with type-specific primers were used according to the modified protocol used by Rose et al.11
For detection of G238C transversion; 4.5 µl of genomic DNA was amplified either with 4 µl of primer: P2WF (5’-GTATGATTTTATGCAGGTTTG-3’) for wild type or P2M (5-GTATGAATTTTATGCAGGTTTC-3 for mutant one, four µl of primer P2C (5-TAAATAGGAACCATCGGACAC-3 was added to each amplification. To detect the G460A transition, a second PCR assay was done by using 4 µl of primers P460F (5-AGGGAGCTAGGGAAAAAGAAAGGTG-3) and P460R (5- CAAGCCTTATAGCCTTACACCCAGG-3). For RFLP, the PCR product was digested with Mwol (restriction enzyme, Fermentas, USA) for one hour at 60°C. To detect the A719G transition, a third PCR assay using primers, P719F (5-GAGACAGAGTTTCACCATCTTGG-3) and P719R (5- CAGGCTTTAGCATAATTTTCAATTCCTC-3 was done under the same conditions as for those used for 460 transition except that we digested the PCR product with AccI (restriction enzyme, Fermentas, USA).
Electrophoresis on 2% agarose gel was used to observe the PCR and RFLP products stained with ethidium bromide (Figure 1, 2) contamination was avoided by using standard precautionary measures. Statistical analysis was done by using percentages for both quantitative and qualitative variables.
For the detection of G238C, a DNA fragment was amplified with P2M P2C when the C23A mutant was present at 250bp, whereas the DNA fragment was amplified at 250bp with P2W and P2C primers when G238 (wild type) was present (Figure 1 and 2). MwoI digestion of wild type was done for deletion of G468 transition which yielded fragments of 443 and 251bp whereas DNA containing the G468 transition/mutation is not digested and yields an uncleaved fragment of 694bp (Figure 2). During RFLP of A719G after the introduction of an AccI restriction site in the amplified: wild-type DNA yielded an uncleaved fragment of 373bp while transition/mutation of A719G yielded fragments of 283 and 90 base pairs (Figure 1).
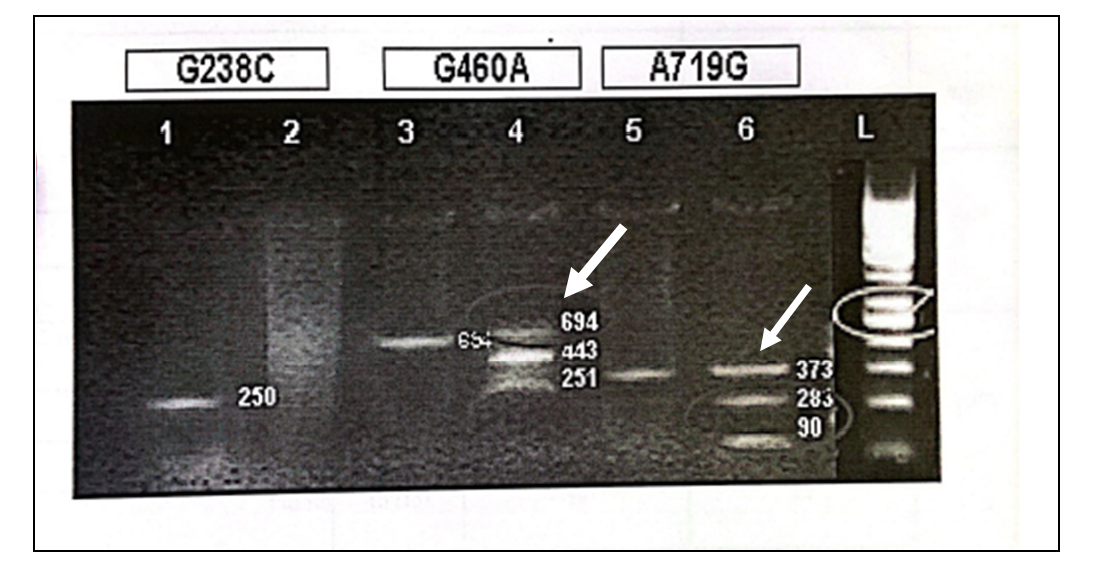
G238C WILDTYPE (250bp)…..POSITIVE
G238C MUTANT TYPE (250bp)…..NEGATIVE
G460A APMLIFIED PRODUCT (694bp)…..POSITIVE
G460A RFLP PRODUCT (694,443 and 251bp)…..CLEAVED PRODUCT (HETEROZYGOUS TRANSITION PRESENT)
A719G APMLIFIED PRODUCT (373bp)……POSITIVE
A719G RFLP PRODUCT (373,238 and 90bp)……CLEAVED PRODUCT (HETEROZYGOUS TRANSITION PRESENT)
Figure 1: RFLP of A719G after the introduction of an AccI restriction site in the amplified: wild-type DNA yielded an uncleaved fragment of 373bp while transition/mutation of A719G yielded fragments of 283 and 90 base pairs.
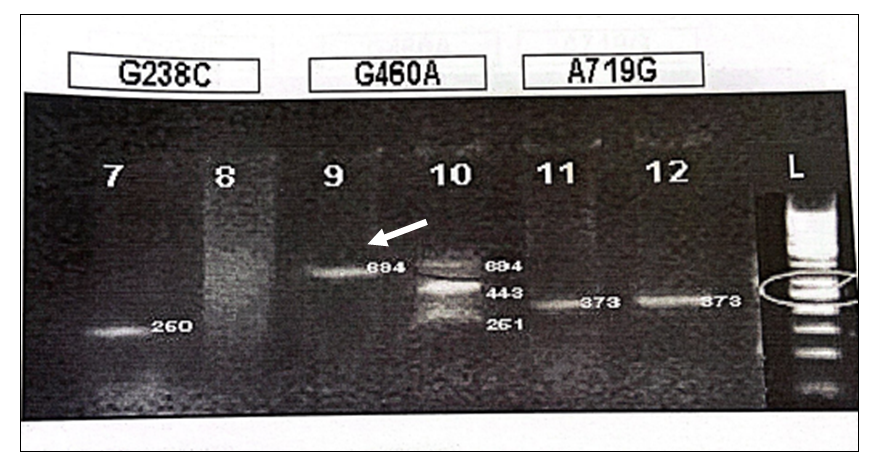
G238C WILDTYPE (260bp)…..POSITIVE
G238C MUTANT TYPE (250bp)…..NEGATIVE
G460A APMLIFIED PRODUCT (694bp)…..POSITIVE
G460A RFLP PRODUCT (694,443 and 251bp)…..CLEAVED PRODUCT (HETEROZYGOUS TRANSITION PRESENT)
A719G APMLIFIED PRODUCT (373bp)……POSITIVE
A719G RFLP PRODUCT (373)……UNCLEAVED PRODUCT (TRANSITION ABSENT)
Figure 2: DNA containing the transition/mutation is not digested and yields an uncleaved fragment of 694bp.
One of the latest studies showed and compared the various pattern of TMPT polymorphic variant alleles in different parts of the world including the study done on South West Asians also which showed the prevalence of TMPT*3A11,12. Many TMPT variant alleles have been detected around the world with many variants reported from one region also. Unlike these reports, only one variant *1/*3B which is very rare in the world was detected in this study as a major component. Keeping in view the variable frequencies of the four common TMPT alleles *2, *3A, *3B and* 3C worldwide this study also analyzed the same TMPT allelic variants. By reviewing the literature on TMPT population studies it was noted that 19 cases of TMPT*3B have been reported from a total of 12208 alleles, a frequency of only 0.16% although their homozygous and heterozygous variants were not identified. Whereas, in this study, 96.94% of patients were identified with heterozygous variant *1/*3B. The polymorphic variant alleles TMPT *3A and TMPT*3C accounted for 4% (488 alleles) and 1.38% (168 alleles), respectively, of the total amount of alleles published while this study showed only 03.06% of patients with heterozygous *1/*3A and none of the patients was found to express *2 or *3C allele.
Table 1: Genotype distribution in the population studied.
| TPMT | Male | Female | Total |
| 56(57.14%) | 42(42.86%) | 98 | |
| *1/*2 | 0 | 0 | 0 |
| *1/*3A | 2 | 1 | 3 |
| *1/*3B | 54 | 4 | 95 |
| *1/*3C | 0 | 0 | 0 |
This is the first study of its kind in Pakistan with unexpected genotyping results of TMPT variant alleles. Because of these results, it is suggested that children identified as heterozygous *1/*3B TMPT genotype may be started with a lower dose of 6-MP while ALL patients identified as a homozygous variant may be supervised strictly with caution during the period of 6-MP therapy.
To further validate the results and to make it more applicable for adjusting the dosage of 6-MP in childhood ALL in Pakistan, it is suggested that large population-based studies of TMPT genotyping of ALL patients as well as of healthy individuals may be done. This can be approached through advanced or alternative technologies analyzing the role of TMPT genetic polymorphism including pyro-sequencing of the TMPT gene as well as HPLC of drug metabolite levels of 6-MP. Day-to-day improvements and advancements in pharmacogenetics and human genomics of different drugs including 6-mercaptopurine worldwide suggest.
Table 2: Demographic data of ALL patients positive for *1/*3B genotype.
| Variables | Frequency n (%) | |||||
| Gender | Male | Female | ||||
| 54(56) | 41(43.2) | |||||
| Age Group | 1.1 – 5 years | 5 – 10 years | 10.1 years – onwards | |||
| 40(42.1) | 31(32.6) | 24(25.3) | ||||
| Ethnicity | Urdu | Sindhi | Balochi | Punjabi | Pushto | Hindu |
| 35(36.8) | 31(32.6) | 8(8.42) | 7(7.37) | 13(13.7) | 1(1.05) | |
| Family History of Cancer | Yes | No | ||||
| 10(10.5) | 85(89.5) | |||||
| Consanguinity | Yes | No | ||||
| 52(54.7) | 43(45.3) | |||||
| Socioeconomic Status – Income | Lower | Middle | High | |||
| 72(75.8) | 21(22.1) | 2(2.11) | ||||
The current study presented data on the evaluation of the genetic polymorphism of Thiopurine S-methyltransferase (TPMT) in 100 children of different ages with acute lymphoblastic leukemia (ALL). The obtained frequency characteristics of the occurrence of TRMT genotypes: *1/2 – 0.00%, *3A – 3.06%, *3B – 96.94% and *3C – 0.00% are comparable with published literature data. We analyzed the relationship of TPMT genetic polymorphism with the toxicity of treatment in the study. According to the ALL-MB-2002 protocol13, a significant predominance of severe hematological toxicity in heterozygous carriers of mutant alleles was established in comparison with carriers of the “wild” type. An increase in the duration of severe myelotoxicity or myelosuppression at the stages of consolidation with TPMT genetic polymorphism was also established.
According to Ando et al. Mercaptopurine (6-MP) is the main component of most chemotherapy regimens in the maintenance phase of acute lymphocytic leukemia (ALL) treatment14. Thiopurine methyltransferase (TPMT) is one of the enzymes responsible for the metabolism of 6-MP. Several studies demonstrate a co-dominant autosomal genetic polymorphism for TPMT in humans hence, 6-MP metabolism is intensely affected by this polymorphism14,15.
In this study, high intra-erythrocyte concentrations of 6-tioguanine nucleotides(6-TGN) and low concentrations of methylmercaptopurine (MMP) were found. Therefore, it was not possible to carry out statistical analyzes to evaluate the relationship between the genotype and the concentration of 6-MP metabolites. Our results in Pakistani child patients shared similarities with Silva et al. in which the TPMT*3A was the second most frequent allele in the Brazilian population15. Several indications found in the present study suggest that the intensity of chemotherapy may not have been sufficient, especially in the group of patients with TPMT gene mutation. This would explain why no significant differences were found in the prognosis of the groups with and without TPMT gene mutation. The incorporation of genotyping into the routine follow-up of patients treated could be an auxiliary tool in the optimization of 6-MP doses, minimizing the risks of toxicity and contributing to a better diagnostic and therapeutic approach16.
However, Alsous et al. observed severe bone marrow toxicity. According to the dose of 6-MP when the patient presents toxicity grades 3 or 4 attributable to this drug. However, it was not possible in our study that toxicity is due to 6-MP since patients receive polychemotherapy and it could not either assume it was due to variant genotypes of the gene TPMT 16. Similarly, Kapoor et al. reported that a common practice is to decrease the dose of more than one drug and this reduction does not eliminate the risk of serious toxicity to 6-MP in patients with TPMT deficiency. If it has done a reduction in the dose of 6-MP, we must be aware that this action may impact the intracellular concentrations of TGNs, reducing the effectiveness of the drug and increasing the risk of relapse. In addition to the fact that the use of sub-therapeutic doses may explain at least in part of the development of resistance to antineoplastic agents17.
It should also be considered that there can be several factors that determine toxicity to 6MP, including decreased excretion rate, the interaction with other drugs, or polymorphisms in different proteins involved in its metabolism or transport. The identification of the genotype of TPMT is an objective element that allows defining the doses of thiopurines individually even before starting the treatment. This would reduce the risk of severe hematological toxicity and would favorably modify the morbidity and survival of patients who require these drugs18. The PCR-RFLP and allele-specific PCR techniques are relatively simple and do not involve a high cost. In emerging economies, many centers that were developed for investigating gene polymorphisms using PCR-based methods face issues due to the implementation of DNA sequencing technologies16. However, in our setting, it is undeniable and PCR techniques can be easily and widely applied at a low cost in a basic molecular biology laboratory.
The analysis of the gene polymorphisms in TPMT is important in the evaluation of patients who require thiopurines as a part of the treatment and represents one of the best examples of pharmacogenetic studies19,13. In some centers, this analysis is carried out as a routine to individualize the doses of these medications. Individual variation in the ability of TPMT is determined by the genotype resulting from paternal and maternal alleles that each inherits19. According to Mcleod et al., the decrease or loss of this activity gives rise to an increase in the active metabolites of thiopurines, resulting in enhanced pharmacological action followed by the adverse effects of these drugs20.
Differences in frequency were well demonstrated in our study. The frequency of the variant TPMT*3B has not been evaluated significantly. However, some studies carried out in Latin America showed higher allele frequency of TPMT*3A in ALL patients from Argentina21. In the Asian population, the variant TPMT *3C was the most frequent22. In these studies, there was a high frequency of individuals carrying variant alleles, with a major frequent percentage analyzed for TPMT*3C. It contrasted with what has been described in populations, like Caucasian, Asian or African, in which there was a clear predominance of some variants and none of them reported a higher frequency of the variant23, 24. TPMT*3B did not follow any pattern of those reported in previous studies; in fact, there was no clear predominance of no variant and the difference between them. In one of them, it was one to two cases. Nevertheless, we believe that these data should be considered when studying the genotype of the enzyme TPMT with a large Pakistani population.
Similarly, an understanding of the molecular mechanisms should be based on interindividual differences in chemotherapy drugs’ effects. It helps in the personalization of treatment, improving the results of treatment of acute leukemia and reducing its toxicity25. Certainly, the metabolic process is complex, and evaluating the contribution of each of the enzymes to concentration levels of active metabolites along with its efficacy and toxicity of therapy is not an easy one. However, despite this, for over the past 10 years, researchers have made significant progress in understanding the reasons for differences in efficiency and tolerability of 6-MP, resulting in practical recommendations for individual doses depending on the genotypes of a metabolic enzyme, TPMT20, 22.
Not all cases exhibit toxicity by mutations. However, using this approach can help to avoid the development of severe myelosuppression, and at the same time, to prevent infectious complications thus, reduce the cost of unnecessary hospitalizations and indirectly increasing the effectiveness of therapy giving the elimination of interruptions in the reception of 6-MP in several patients. Personalization of chemotherapy is one of the priority areas for the development of onco-hematology26.
All the patients in the study group were found to have heterozygous TPMT polymorphic alleles. Out of them, 74.5% of ALL patients showed myelotoxicity. Furthermore, *1/*3B was the most prevalent variant TPMT allele observed with the highest frequency reported at 97%. More research is required relating to pharmacogenetics so that researchers can understand the number of drugs, the creation of panels of genes, and determine the mutations before the start of therapy. In the future, this may change the approach to the treatment of onco-hematological diseases. As a result, it will be possible to achieve maximizing the effectiveness of treatment with minimizing systemic toxicity.
The authors would like to acknowledge the hospital staff for their assistance and facilitation in the data collection process.
The authors declared no conflict of interest.
The study approval was obtained by the institutional ethics review board.
Informed consent was taken from the patients.
All authors equally contributed to this research study.
- Rivera GK, Pinkel D, Simone JV, Hancock ML, Crist WM. Treatment of acute lymphoblastic leukemia–30 years’ experience at St. Jude children’s research hospital. N Engl J Med. 1993;329(18):1289-1295. doi: 10.1056/NEJM199310283291801
- Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535-1548. doi: 10.1056/NEJMra023001
- Cannon JG. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. Edited by Laurence Brunton, John Lazo, and Keith Parker. McGraw Hill, New York. 2005, p. 1414.
- Tai HL, Krynetski EY, Yates CR, Loennechen T, Fessing MY, Krynetskaia NF, et al. Thiopurine S-methyltransferase deficiency: two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am J Hum Genet. 1996;58(4):694-702.
- Krynetski EY, Evans WE. Genetic polymorphism of thiopurine S-methyltransferase: molecular mechanisms and clinical importance. Pharmacology. 2000;61(3):136-146. doi: 10.1159/000028394
- Weinshilboum R. Thiopurine pharmacogenetics: clinical and molecular studies of thiopurine methyltransferase. Drug Metab Dispos. 2001;29(4):601-605.
- Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43(4):329-339. doi: 10.1007/BF02220605
- Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32(5):651-662.
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371(9617):1030-1043. doi: 10.1016/S0140-6736(08)60457-2
- McLeod HL, Krynetski EY, Relling MV, Evans WE. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia. 2000;14(4):567-572. doi: 10.1038/sj.leu.2401723
- Rose CM, Marsh S, Ameyaw MM, McLeod HL. Pharmacogenetic analysis of clinically relevant genetic polymorphisms. Novel Anticancer Drug Protoc. 2003:225-237. doi: 10.1385/1-59259-380-1:225
- Chrzanowska M, Kuehn M, Januszkiewicz-Lewandowska D, Kurzawski M, Droździk M. Thiopurine S-methyltransferase phenotype-genotype correlation in children with acute lymphoblastic leukemia. Acta Pol Pharm. 2012;69(3):405-410.
- Rumyantseva YV, Karachunsky AI, Aleinikova OV, Fechina LG, Shamardina AV, Litvinov DV, et al. Efficiency of the ALL-MB-2002 protocol in children with acute lymphoblastic leukemia. Ter Arkh. 2010;82(7):11-19.
- Ando M, Ando Y, Hasegawa Y, Sekido Y, Shimokata K, Horibe K. Genetic polymorphisms of thiopurine S-methyltransferase and 6-mercaptopurine toxicity in Japanese children with acute lymphoblastic leukaemia. Pharmacogenet Genomics. 2001;11(3):269-273.
- Silva MR, de Oliveira BM, Viana MB, Murao M, Romanha AJ. Thiopurine S-methyltransferase (TPMT) gene polymorphism in Brazilian children with acute lymphoblastic leukemia: association with clinical and laboratory data. Ther Drug Monit. 2008;30(6):700-704. doi: 10.1097/FTD.0b013e31818b0f31
- Alsous M, Yousef AM, Jalil MA, Zawiah M, Yacoub S, Momani D, et al. Genetic polymorphism of thiopurine S-methyltransferase in children with acute lymphoblastic leukemia in Jordan. Asian Pac J Cancer Prev. 2018;19(1):199-205. doi: 10.22034/APJCP.2018.19.1.199
- Kapoor G, Sinha R, Naithani R, Chandgothia M. Thiopurine S-methyltransferase gene polymorphism and 6-mercaptopurine dose intensity in Indian children with acute lymphoblastic leukemia. Leuk Res. 2010;34(8):1023-1026. doi: 10.1016/j.leukres.2010.01.029
- Farfan MJ, Salas C, Canales C, Silva F, Villarroel M, Kopp K, et al. Prevalence of TPMT and ITPA gene polymorphisms and effect on mercaptopurine dosage in Chilean children with acute lymphoblastic leukemia. BMC Cancer. 2014;14(1):1-5. doi: 10.1186/1471-2407-14-299
- McLeod HL, Relling MV, Liu Q, Pui CH, Evans WE. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood.1995;85(7):1897-1902. doi: 10.1182/blood.V85.7.1897.bloodjournal8571897
- McLeod HL, Coulthard S, Thomas AE, Pritchard SC, King DJ, Richards SM, et al. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol. 1999;105(3):696-700. doi: 10.1046/j.1365-2141.1999.01416.x
- Aráoz HV, D’Aloi K, Foncuberta ME, Sanchez La Rosa CG, Alonso CN, Chertkoff L, et al. Pharmacogenetic studies in children with acute lymphoblastic leukemia in Argentina. Leuk Lymphoma. 2015;56(5):1370-1378. doi: 10.3109/10428194.2014.951844
- Kim HY, Lee SH, Lee MN, Kim JW, Kim YH, Kim MJ, et al. Complete sequence-based screening of TPMT variants in the Korean population. Pharmacogenet Genomics. 2015;25(3):143-146. doi: 10.1097/FPC.0000000000000117
- Engen RM, Marsh S, Van Booven DJ, McLeod HL. Ethnic differences in pharmacogenetically relevant genes. Curr Drug Deliv. 2006;7(12):1641-1648. doi: 10.2174/138945006779025446
- Marinaki AM, Arenas M, Khan ZH, Lewis CM, Shobowale-Bakre EM, Escuredo E, et al. Genetic determinants of the thiopurine methyltransferase intermediate activity phenotype in British Asians and Caucasians. Pharmacogenet Genomics. 2003;13(2):97-105.
- Rather RA, Bhagat M. Cancer chemoprevention and piperine: molecular mechanisms and therapeutic opportunities. Front Cell Dev Biol. 2018;6:1-12. doi: 10.3389/fcell.2018.00010
- Badalian-Very G. Personalized medicine in hematology—A landmark from bench to bed. Comput Struct Biotechnol J. 2014;10(17):70-77. doi: 10.1016/j.csbj.2014.08.002
This is an open-access article distributed under the terms of the CreativeCommons Attribution License (CC BY) 4.0 https://creativecommons.org/licenses/by/4.0/